



















به آرین درمان پژوه خوش آمدید
شرکت مهندسی آرین درمان پژوه با بیش از 15 سال سابقه درخشان در امر تامین تجهیزات تصویربرداری پزشکی مفتخر است در دامنه وسیعی از محصولات با کیفیت از برندهای صاحب نام دنیا نسبت به ارائه خدمت به سامانه درمانی و تشخیص کشور و جامعه محترم پزشکان متخصص رادیولوژی و رادیوگرافی فک و صورت مبادرت ورزد.







700
رادیولوژی دیجیتال
320
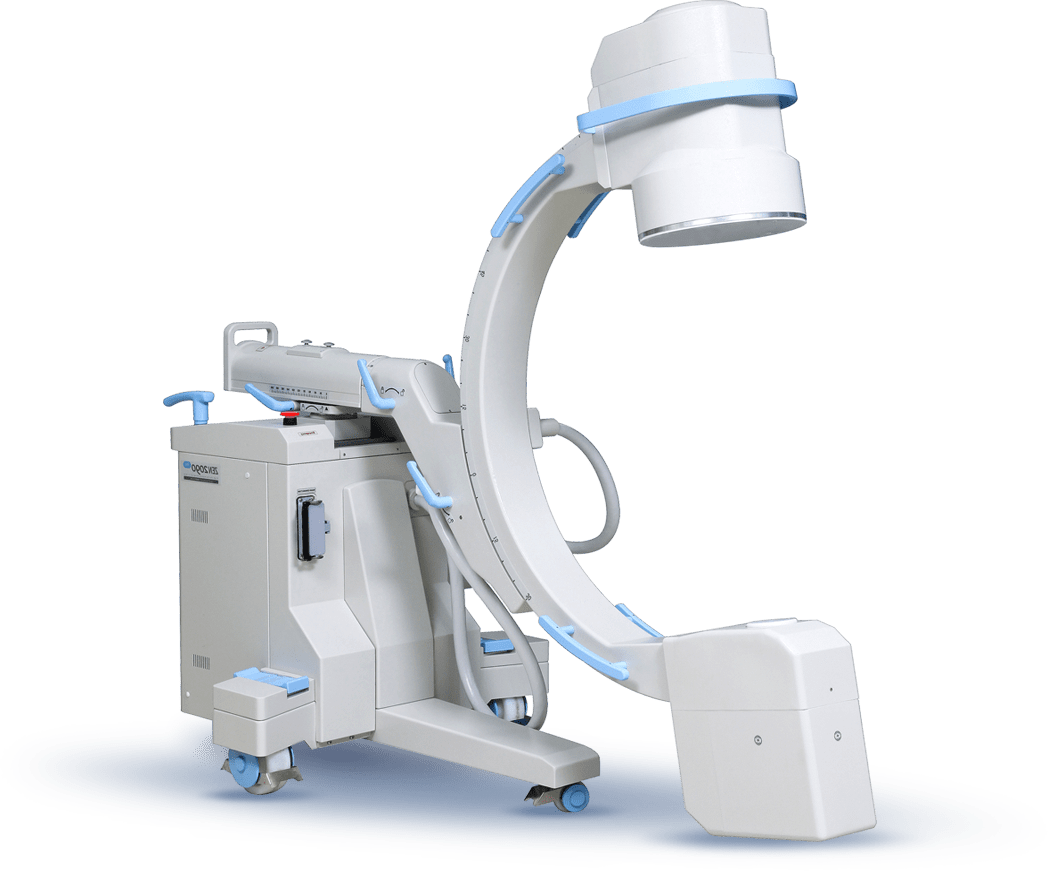
سی آرم و پرتابل
1200
OPG
40
CBCT
6000
سنسور RVG
1200
PSP و PA
















03
اسفند
گردهمایی انجمنهای گردشگری سلامت در دبی
گردهمایی انجمنهای گردشگری سلامت و شرکتهای گردشگری درمانی با مدیریت مرکز توسعه گردشگری سلامت کشورهای اسلامی و انجمن گردشگری سلامت استا...

03
اسفند
قراردادهای ۳.۹ میلیارد دلاری در نمایشگاه و کنفرانس دندانپزشکی دبی
بخشی از نمایشگاه دندانپزشکی دبی (AEEDC) که اخیرا پایان یافت.
دبی یکشنبه (۶ فوریه) اعلام کرد ارزش قراردادهای انعقادی مستقیم و غیر مستقی...

18
فروردین
نمایشگاه KIMES 2021
'KIMES 2021' ، بزرگترین نمایشگاه صنعت پزشکی کره با موفقیت به پایان رسید. کمپانی Genoray از بازدیدکنندگان زیادی که علی رغم شرایط دشوار...
راه های تماس 
برای برقراری تماس با ما راه های مختلفی پیش روی شماست. فقط کافیست یکی را انتخاب کنید و سؤالات ، پیشنهادات و انتقادات خود را برای ما ارسال کنید. خیلی سریع پاسخگو هستیم.
آدرس
ساعات کاری
شماره های تماس
ایمیل
شبکه های اجتماعی